


阳极是电解海水制氯防污装置中的关键材料[1,3~5],要求具有高的析氯活性和长的使用寿命。钛基金属氧化物阳极,也称为尺寸稳定性阳极(DSA),是将导电的金属氧化物涂覆在金属钛基体上制成的一系列电极[3]。早期的DSA主要为钛基RuO2-TiO2涂层阳极,该阳极和石墨阳极相比,具有电化学活性高、消耗速率小、尺寸稳定等优点,在氯碱工业上得到广泛使用[3,4]。然而,尽管RuO2-TiO2涂层阳极在电解饱和浓盐水时具有优良的性能,但在电解稀盐水或海水条件下使用寿命却很短,难以满足实际工程的需要[4,5]。为此,发展了钛基RuO2-IrO2-TiO2涂层阳极,通过加入高稳定性的活性组元IrO2,可明显改善RuO2-TiO2氧化物阳极的稳定性[6~10]。
RuO2-TiO2电极在使用过程中的失效主要归结为[8,11]:(1) 涂层活性组元的溶解消耗;(2) 涂层下的钛基体发生钝化,形成绝缘的TiO2钝化膜;(3) 局部涂层在析出气体的冲刷下发生脱落。因此,要提高金属氧化物阳极的使用寿命,除了改善金属氧化物涂层本身的稳定性以外,还需要改善氧化物电催化涂层和钛基体之间的界面性能。钛基体的预处理及表面状态是影响钛基金属氧化物阳极性能的主要因素之一。初立英等[12]研究表明,钛基体喷砂处理后在草酸中的刻蚀时间对氧化物阳极的性能有显著影响,只有获得合适的多孔钛基体表面才能够提高氧化物阳极的电催化活性和稳定性。有研究表明[13],通过在钛表面形成TiH2氢化物层可以改善氧化物涂层和基体间界面的导电性能,降低电催化涂层和钛基体之间的欧姆降。然而,关于氢化物层对金属氧化物阳极性能的影响仍存在争议。例如,Shao等[14]通过在钛基体上先热分解制备TiO2,然后进行阴极还原的方法制备了含TiH x 中间层的Ti/Sb-SnO2电极,结果表明,含氢化物中间层的电极具有更高的稳定性和更高的氧气析出电位(更低的析氧活性)。而Wu等[15]研究则表明,通过引入TiH2中间层制备的Ti/TiH2/β-PbO2-Mn3O4@C电极具有高的析氧电催化活性,并将其电化学活性的提高归因于低电阻和多孔的TiH2中间层加速了电荷转移速率。因此,不同方法制备的TiH2以及对于不同的氧化物涂层体系,TiH2对氧化物阳极的性能存在不同的影响。
本文采用恒电流阴极充氢方法在钛基体表面生成TiH x,然后采用聚合物溶胶-凝胶法在处理后的钛基体上制备了RuO2-IrO2-TiO2涂层,系统研究了钛基体经不同电流密度的阴极充氢处理后的表面状态,及其对RuO2-IrO2-TiO2阳极微观结构和性能的影响。
1 实验方法
1.1 电极制备
基体材料为工业纯钛(TA2)板,试样尺寸为10 mm × 10 mm × 1.5 mm。钛基体先进行喷砂处理,然后置于10% (质量分数)草酸溶液中于90 ℃刻蚀1.5 h,以除去表面氧化膜,同时增加表面粗糙度。经酸洗后的钛基体呈灰色麻面,用超纯水和无水乙醇进行冲洗,浸泡在无水乙醇中备用。
对酸洗处理后的试样于室温下在0.5 mol/L H2SO4溶液中进行恒电流阴极充氢,采用三电极体系,试样为工作电极、饱和甘汞电极作为参比电极、铂网作为辅助阳极。阴极充氢电流密度分别为50、100、250、500 mA/cm2,极化时间为4 h。经阴极充氢处理后的试样用蒸馏水和酒精清洗,以备后续制备金属氧化物涂层。
采取聚合物溶胶-凝胶法制备RuO2-IrO2-TiO2涂层。将柠檬酸(CA)和乙二醇(EG)加入到三口烧瓶中加热至65 ℃反应30 min,随后按照Ru∶Ir∶Ti = 15∶15∶70的摩尔比向其加入RuCl3、氯铱酸和TiCl4,升温至90 ℃反应2 h,待溶液冷却后,加入正丁醇溶剂,将金属离子浓度调整为0.5 mol/L,即得到制备氧化物涂层阳极所需前驱体涂液。
用毛刷蘸取涂液均匀涂覆在仅经过酸蚀和经不同阴极充氢处理后的钛基体表面,将涂好的钛片放入烘箱中120 ℃干燥10 min,随后放入管式炉中于450 ℃烧结15 min。重复涂覆、烘干和烧结过程5次,最后一次的烧结时间为1 h,即得到所需的氧化物涂层阳极试样。
1.2 结构表征
采用场发射扫描电子显微镜 (SEM,ULTRA55) 观察经预处理钛基体以及金属氧化物阳极试样的表面形貌,操作电压为15 kV。使用X射线衍射仪(XRD,D-MAX2550)对充氢预处理的钛基体以及制备的氧化物涂层阳极进行结构分析,使用Cu Kα射线(λ = 0.1540 nm),扫描速率为5 (°)/min,扫描范围为10°∼90°。
1.3 电化学测试
对所制备的充氢钛基体和氧化物阳极进行电化学测试,使用的设备为多通道电化学测试系统(Biologic VMP3)。电化学测试均在由三电极体系构成的电解池中进行,试样为工作电极,暴露的测试工作面积为1 cm2;铂片为对电极,饱和甘汞电极(SCE)为参比电极。测试介质为室温条件下的3.5% (质量分数) NaCl溶液。文中给出的所有电位均相对于SCE。测试了经不同预处理的钛基体和制备的氧化物阳极的动电位极化曲线,扫描速率为0.33 mV/s,充氢钛基体的电位扫描范围为-1.0~4.0 V (相对于开路电位),氧化物阳极的电位扫描范围为0~2 V (相对于SCE)。对充氢钛基体和氧化物阳极进行了电化学阻抗谱测试,施加电位分别为开路电位(OCP)和1.20 V,交流扰动信号的幅值为10 mV,频率的扫描范围为105~10-2 Hz。对氧化物阳极进行了循环伏安测试,扫描速率分别为10、20、40、60、80、100和120 mV/s,扫描电位范围为0~1.0 V (相对于SCE)。
1.4 加速寿命试验
通过恒电流加速寿命试验来评价金属氧化物阳极的稳定性。加速寿命试验在1 mol/L H2SO4溶液中进行,电解电流密度为1 A/cm²。金属氧化物电极作为阳极,钛板作为阴极。使用TP700数据采集仪记录槽压随时间的变化,当槽压达到10 V即认为氧化物阳极失效,终止试验。此时累计的电解时间即为金属氧化物阳极的强化电解寿命。
2 结果与分析
2.1 阴极充氢对钛基体表面状态的影响
2.1.1 钛基体表面形貌和物相分析
图1

图1
钛基体经酸蚀以及在硫酸溶液中于不同电流密度阴极充氢4 h后的表面形貌
Fig.1
Surface morphologies of titanium substrates with acid etching before (a, b) and after hydrogen charging for 4 h in sulfuric acid solution at the current densities of 50 (c, d), 100 (e, f), 250 (g, h) and 500 (i, j) mA/cm2
当阴极充氢电流密度较低时,钛基体表面形貌变化不太明显。当电流密度增加到100 mA/cm2以上时,试样表面出现暗斑并逐渐增加,面积逐渐扩大,表明表面形成新的产物。当阴极充氢电流密度达到500 mA/cm2时,如图1j所示,试样表面的产物以及该产物与基体的界面处发生了明显的开裂。
对经酸蚀以及在硫酸溶液中于不同电流密度阴极充氢后的钛基体进行了XRD分析,其衍射谱如图2所示。经酸蚀后的钛基体试样呈现出金属α-Ti和TiH1.5 (JCPDS No. 01-078-2216)的衍射峰,TiH1.5的出现应该是在草酸酸洗过程中形成的。该TiH1.5为δ相,具有面心立方结构[16]。当进行阴极充氢后,除了金属Ti和TiH1.5的衍射峰以外,在35.9°检测到了TiH2的特征峰,该氢化物为ε相,具有面心四方结构。随着充氢电流密度的增大,TiH1.5和TiH2的衍射峰逐渐增强,表明由于表面渗入氢含量的增加导致更多的TiH1.5和TiH2生成。同时,钛基体的衍射峰则随充氢电流增加而逐渐减弱,这也表明表面的氢化物层在逐渐增厚。
图2

图2
钛基体经酸蚀以及在硫酸溶液中于不同电流密度阴极充氢后的XRD谱图
Fig.2
XRD spectra of titanium substrates with acid etching before and after hydrogen charging at different cathode current densities in sulfuric acid solution
2.1.2 钛基体表面电化学特性
对经过不同充氢处理的钛基体进行了EIS测量,图3为未充氢和经不同电流密度充氢后的钛基体在3.5%NaCl溶液中于OCP电位下的电化学阻抗谱。由图3a可见,所有钛基体试样的Nyquist图都显示为容抗弧。和没有阴极充氢的试样相比,充氢后钛基体的容抗弧尺寸明显变小。随着阴极充氢电流密度的增加,容抗弧的半径逐渐减小,这表明钛基体耐腐蚀性能的降低。由图3b中的相角图可见,未阴极充氢的试样具有最大的相位角且在很宽的频率范围内都具有较高的相位角,表明钛基体尽管经过草酸刻蚀处理以去除表面氧化膜,但Ti表面极易发生氧化,在NaCl溶液中其表面又形成了较致密TiO2钝化膜[19]。但当充氢电流密度逐渐增加时,试样的最大相位角以及低频处的相位角逐渐下降,表明钛基体表面膜的绝缘性能逐渐降低[19]。由图3c可以看出,未充氢试样呈现出类似纯电容的响应特征,且具有最大的低频(f = 0.01 Hz)阻抗模值。随着充氢电流密度的增加,钛基体试样的低频阻抗模值逐渐减小,这意味着表面膜中的缺陷密度增强,表面膜的完整性减弱[20,21]。
图3
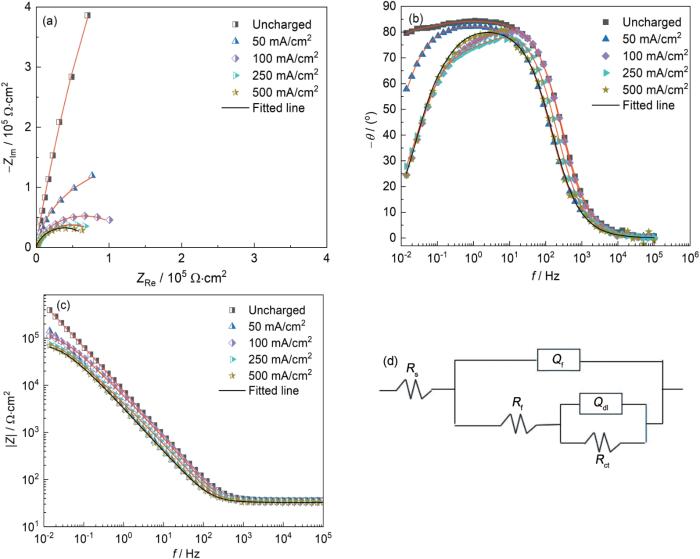
图3
未充氢和经不同电流密度充氢后的钛基体在3.5%NaCl溶液中的电化学阻抗谱
Fig.3
Nyquist (a), Bode phase angle (b) and Bode modulus (c) diagrams of titanium substrates before and after hydrogen charging at different cathode current densities in 3.5%NaCl solution, and corresponding equivalent circuit diagram (d)
其中,ZCPE表示CPE的阻抗,Y0表示模量,ω和n分别为角频率和弥散系数;n通常在0.5~1之间。当n = 1时,CPE为理想电容,当0.5 < n < 1时,CPE为非理想电容。基于上述等效电路模型拟合得到的参数如表1所列。
表1 未充氢和经不同电流密度充氢钛基体的EIS拟合参数
Table 1
| I / mA·cm-2 | Rs / Ω·cm2 | Qf / μΩ·cm2·S n | n1 | Rf / kΩ·cm2 | Qct / µΩ·cm2·S n | n2 | Rct / kΩ·cm2 |
|---|---|---|---|---|---|---|---|
| Uncharged | 36.0 | 23.0 | 0.95 | 962 | 3.03 | 0.88 | 2970 |
| 50 | 34.0 | 30.5 | 0.94 | 63.9 | 12.7 | 0.73 | 150 |
| 100 | 33.9 | 32.7 | 0.92 | 14.8 | 13.7 | 0.71 | 108 |
| 250 | 33.5 | 36.3 | 0.88 | 8.09 | 16.5 | 0.74 | 91.1 |
| 500 | 34.5 | 38.9 | 0.93 | 5.51 | 17.3 | 0.70 | 63.9 |
从表1可知,未充氢试样的膜电阻Rf为962 kΩ·cm2,而经50 mA/cm2阴极电流密度充氢后,表面膜电阻为63.9 kΩ·cm2,降低一个数量级,表明阴极充氢明显提高了表面膜的电导率。随着充氢电流密度的加大,Rf值继续降低,当充氢电流密度为500 mA/cm2时,Rf降低到5.51 kΩ·cm2。该结果表明,未充氢试样尽管进行了酸蚀处理,其表面仍存在薄的氧化膜。有研究表明[20],未充氢TA2试样表面膜的成分主要为TiO2,阴极充氢后表面膜中TiO2含量会明显降低,而低价态的氧化物Ti2O3、TiO的含量则增加。这种氧化膜成分的变化会导致膜电阻降低。随着充氢电流密度的增大,低价态的氧化物进一步增多,同时有更多Ti的氢化物生成,结果导致了膜电阻的进一步降低。从Rct的变化来看,和未充氢试样相比,充氢试样的Rct显著降低,并且随着充氢电流密度的增加,Rct的值继续减小。Rct反映了表面膜孔隙、裂纹等缺陷处的钛基体的电化学反应活性,其值越小,表明试样的耐腐蚀性能越低,表面膜对TA2基体的有效保护作用越弱。此外,阴极充氢导致氢化物的析出和增多不仅降低了表面膜的保护作用,而且由于体积变化会引起钛基体局部应变增加和表面裂纹产生,从而导致耐腐蚀性能的降低[20]。因此,阴极充氢降低了钛基体的耐蚀性能。膜电容和双电层电容也是反映表面电化学特性的重要参数,阴极充氢后,试样的膜电容和双电层电容均明显增大,表明膜层的介电性能降低(因含有更多低价态氧化物和氢化物),膜层存在更多缺陷,导致膜层下更大面积钛基体和溶液接触,参与电化学反应。EIS测试的结果和前面表面形貌和相分析结果是一致的。
图4
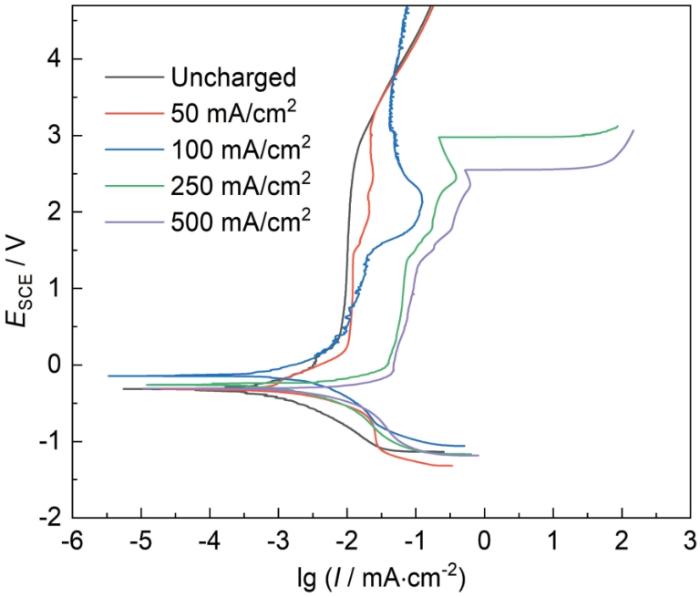
图4
未充氢和经不同电流密度充氢钛基体的动电位极化曲线
Fig.4
Potentiodynamic polarization curves of titanium substrates uncharged and charged with hydrogen at different cathode current densities
未充氢试样具有较低的自腐蚀电流密度和明显的自钝化特征,没有活化-钝化过程,表明试样表面有氧化膜存在,可对基体有较好的保护作用。对于充氢试样,阳极极化曲线也呈现出钝化特征,但在电位达到大约1.5 V左右时,电流密度会明显增大,出现过钝化,而随着电位进一步正移,电流密度又逐渐降低,低电流密度(50和100 mA/cm2)充氢试样的极化曲线逐渐和未充氢试样重合,而大电流密度(250和500 mA/cm2)充氢试样则出现了表面膜击穿现象,电流密度显著增大,表明钛基体发生了点蚀。此外,和未充氢试样相比,充氢试样的钝化电流密度、过钝化区的电流密度峰值都更大,且随着充氢电流密度的升高而增大,表明表面膜层具有更低的保护作用。充氢试样表面含有低价氧化物和氢化物,在高电位下会发生氧化反应,过钝化区的出现可能与表面膜的成分和结构发生转变有关[20,22]。在高电流密度下充氢的试样,由于具有更高的氢含量和更多的氢化物形成,其表面膜存在更多缺陷,钝化膜的稳定性降低,因而击穿电位更低,更易于发生钛基体点蚀。考虑到金属氧化物阳极采用阀金属钛做基体,是因为当氧化物涂层发生局部损坏时,裸露的钛基体因表面形成钝化膜而得到保护。Ti在海水中的钝化膜击穿电位大约为8~12 V[1],因此当采用充氢预处理的钛做金属氧化物阳极的基体时,则会因其击穿电位的明显降低而显著增大金属氧化物阳极失效的风险。
2.2 基体阴极充氢对RuO2-IrO2-TiO2 阳极的影响
2.2.1 氧化物阳极的微观结构
图5为在未充氢的钛基体以及经不同电流密度充氢钛基体上制备的氧化物阳极的表面形貌。所有氧化物阳极表面均呈现出热分解法制备氧化物阳极所具有的典型的泥裂结构和深浅不一的凹坑。凹坑是因为在基体预处理的过程中草酸刻蚀留下的,因为氧化物涂层本身的厚度较小,所以基体本身的粗糙结构也会在所制备的氧化物涂层阳极上得以呈现。泥裂结构的形成是由于在氧化物阳极制备过程中需反复进行加热和冷却处理,且由于基体与涂层的热膨胀系数不同而导致氧化物涂层存在较大内应力所致。随着充氢电流密度逐渐增加,大裂纹逐渐增多并且出现大裂块,当充氢电流密度为250 mA/cm2时,表面形貌主要由大裂块组成,大而深的裂纹最多并形成网络结构。然而,当电流密度达到500 mA/cm2时,表面粗大的裂纹却明显减少,表面主要由较小的裂块组成。
图5

图5
钛基体经酸蚀以及不同电流密度阴极充氢后制备的RuO2-IrO2-TiO2阳极的表面形貌
Fig.5
Surface morphologies of RuO2-IrO2-TiO2 anodes prepared on acid-etched titanium substrates uncharged (a) and charged with hydrogen at the cathode current densities 50 (b), 100 (c), 250 (d) and 500 (e) mA/cm2
氧化物涂层表面裂纹形貌受到很多因素的影响,由于溶胶凝胶法所采用的前驱体涂液通常黏度较大,因此每次的涂膜较厚,热烧结制备氧化物阳极过程中存在更大的涂层内应力,故氧化物阳极出现大量的微裂纹。此外,涂层的裂纹还受到基体表面状态以及上道涂层表面形貌的影响。和未充氢钛基体相比,随着充氢电流密度的增大,更多的氢化物在表面生成,导致表面不均匀性增加(图1),有利于粗大裂纹的形成。而当充氢电流密度为500 mA/cm2时,表面析出的氢化物增多,分布相对均匀,结果导致氧化物涂层表面粗大裂纹又有所减少。
图6为钛基体未充氢和经不同电流密度充氢后制备的氧化物阳极的XRD谱图。由图中可见,存在金红石相的 (Ru, Ir, Ti)O2固溶体和金属Ti的特征峰[7,9]。出现在27.6°、36.0°和54.3°的3个峰对应于金红石相的(110)、(101)和(211)晶面。当烧结温度达到450 ℃以上时IrO2和RuO2可以产生晶红石相,由于Ru、Ir、Ti具有相似的离子半径以及相同的氧化物晶型(金红石相)[23],因此可以形成金属氧化物固溶体。XRD谱图中出现了金属Ti的衍射峰,这是因为X射线穿透了氧化物涂层检测到了钛基体的信息。当充氢电流密度不大于100 mA/cm2时,(Ru, Ir, Ti)O2固溶体特征峰的峰强和位置没有较大的变化,但当电流密度增加到250 mA/cm2及以上时,(Ru, Ir, Ti)O2固溶体特征峰的峰强稍有降低,但其特征峰位置没有较大变化,这可能与TiH x 中间层有关。图6中所有氧化物阳极均没有出现TiH x 的特征峰,这可能是由于烧制阳极温度超过400 ℃,使得TiH x 发生了分解,转变为Ti和H2,如反应式(2)所示[14]。
图6
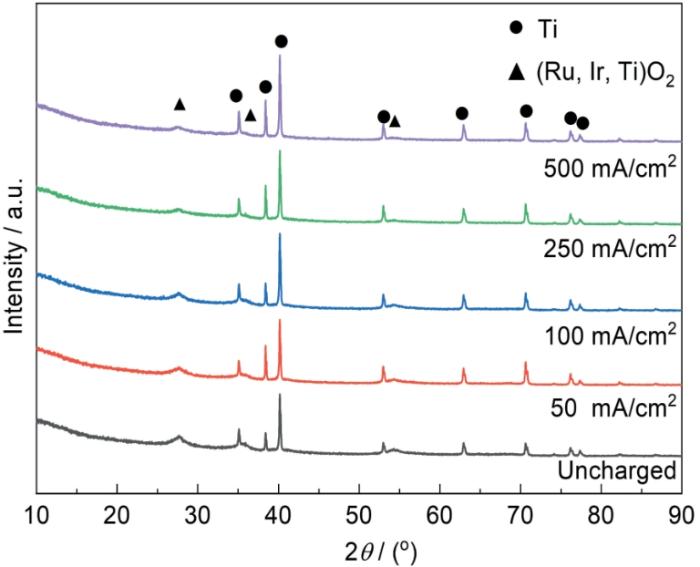
图6
钛基体未充氢和经不同电流密度充氢后制备的RuO2-IrO2-TiO2阳极的XRD谱图
Fig.6
XRD patterns of RuO2-IrO2-TiO2 anodes prepared on titanium substrates uncharged and charged with hydrogen at different cathode current densities
2.2.2 氧化物阳极的电化学性能
为探究钛基体阴极充氢对RuO2-IrO2-TiO2阳极的电化学性能的影响,对制备得到的氧化物阳极在3.5%NaCl溶液中进行了电化学阻抗谱测试,施加的电位为1.20 V (vs.SCE),结果如图7a所示。由Nyquist图可以看出,各阳极的阻抗谱均只有一个容抗弧,钛基体充氢处理的氧化物阳极的容抗弧直径均小于未充氢钛基氧化物阳极,说明钛基体充氢降低了电极阻抗,且随着充氢电流密度的逐渐增加,容抗弧直径也在逐渐减小。但在充氢电流达到500 mA/cm2时,容抗弧直径比250 mA/cm2时有所增加。在施加的电位下阳极表面发生析氯反应,因此容抗弧的大小反映了氧化物阳极析氯电化学反应的活性[23]。阻抗谱在低频区出现明显的扩散尾,这可能是因为EIS测试时氧化物阳极表面积聚的气泡所导致的。还注意到容抗弧为凹陷的半圆,呈现弥散效应,这是典型的多孔电极的特征,表明电极的表面存在较大的粗糙度[8,23]。采用图7b所示等效电路对EIS进行拟合。电路中的Rs表示溶液电阻;Rct为氧化物阳极析氯反应的电荷转移电阻;Qdl是常相位角元件(CPE),其表示受到粗糙表面弥散效应影响的双电层电容;W是Warburg阻抗。通过上述等效电路的模拟得到EIS拟合参数如表2所示。
图7

图7
钛基体未充氢和经不同电流密度充氢后制备的RuO2-IrO2-TiO2阳极在3.5%NaCl溶液中于1.20 V (vs. SCE)测量的电化学阻抗谱和拟合用等效电路图
Fig.7
Nyquist diagrams recorded in 3.5%NaCl solution at 1.20 V (vs. SCE) for RuO2-IrO2-TiO2 anodes prepared on titanium substrates without and with hydrogen charging at different current densities (a), and equivalent circuit diagram (b)
表2 钛基体未充氢和经不同电流密度充氢后制备的氧化物阳极的EIS拟合参数
Table 2
| I / mA·cm-2 | Rs / Ω·cm2 | Qdl / mΩ·cm2·Sn | n | Rct / Ω·cm2 | W / Ω·cm2 |
|---|---|---|---|---|---|
| Uncharged | 32.97 | 9.25 | 0.79 | 12.57 | 3.37 |
| 50 | 31.05 | 10.5 | 0.81 | 10.25 | 1.84 |
| 100 | 29.77 | 10.9 | 0.80 | 7.15 | 2.20 |
| 250 | 31.36 | 19.3 | 0.74 | 4.04 | 1.49 |
| 500 | 31.16 | 15.0 | 0.77 | 4.34 | 1.51 |
从表2可看出,与钛基体未充氢的氧化物阳极相比,钛基体充氢处理后,氧化物阳极的电荷转移电阻Rct均降低,并随着充氢电流密度的增加而减小,但当充氢电流密度达到500 mA/cm2 时,Rct又稍有增大。Rct的减小有利于促进电荷转移,降低表面析氯电化学反应的阻力。这说明钛基体充氢处理会提高阳极的析氯活性,增强氧化物阳极的电催化性能。而Qdl则呈现相反变化趋势,和钛基体未充氢的氧化物阳极相比,钛基体经充氢处理的氧化物阳极的Qdl增大,且随充氢电流密度增大而逐渐增加。当充氢电流密度达到500 mA/cm2时,阳极的Qdl又有所降低。Qdl与氧化物阳极的电化学活性表面积相关,上述结果表明,对钛基体充氢处理可增大氧化物阳极的电化学活性表面积,但当充氢电流密度过大(500 mA/cm2)时,阳极的电化学活性表面积又会减小。钛基体充氢处理后氧化物阳极电化学活性表面积和析氯电催化活性的变化与基体表面钛氢化物层的生成以及氧化物涂层的表面微观结构有关。一方面,充氢所形成的氢化物可以改善金属氧化物涂层和钛基体界面的导电性,有利于提高电荷转移速率,从而提高氧化物阳极的电催化性能[15];另一方面,随充氢电流密度增大,氧化物阳极表面的裂纹,尤其是大而深的裂纹增多(图5),这增大了氧化物阳极的比表面积,使得更多的氧化物活性位点与电解质溶液接触,从而提高了金属氧化物阳极的电化学活性。充氢电流密度为500 mA/cm2时,由于氧化物阳极表面粗大裂纹的减少,导致其电化学活性表面积和电催化活性有所降低。
测试了所制备的氧化物涂层阳极在3.5%NaCl溶液中于不同扫速下的循环伏安曲线,如图8a~e所示。在所选电位区间 (0~1.0 V vs. SCE)主要发生活性组元Ru和Ir不同价态之间的氧化还原转变。表面氧化还原反应与表面活性位点有关,因此,由CV曲线得到的伏安电量q*与活性位点的数量成正比,即与氧化物阳极的实际电化学活性表面积成正比。
图8
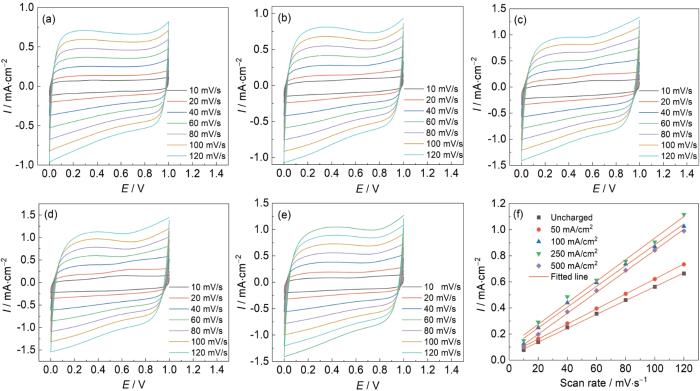
图8
钛基体未充氢和经不同电流密度充氢处理后制备的氧化物阳极在3.5%NaCl溶液中于不同扫速下的循环伏安曲线和0.7 V (vs. SCE)电位下的电流密度与扫描速率的关系曲线
Fig.8
Cyclic voltametric curves recorded in 3.5%NaCl solution at different scanning rates for the oxide anodes on titanium substrates uncharged (a) and charged with hydrogen at the current densities of 50 (b), 100 (c), 250 (d) and 500 (e) mA/cm2, and variations of the current density at 0.7 V (vs. SCE) with scanning rate (f)
将CV曲线的积分面积除以电位扫描速率可计算得到该扫速下的伏安电量q*。可采用式(
表3 钛基体未充氢和经不同电流密度充氢后制备的氧化物阳极的伏安电量参数和孔隙率
Table 3
| I / mA·cm-2 | q | q | q | ε |
|---|---|---|---|---|
| Uncharged | 16.65 | 9.30 | 7.35 | 0.44 |
| 50 | 20.91 | 9.81 | 11.10 | 0.53 |
| 100 | 30.34 | 12.12 | 18.22 | 0.60 |
| 250 | 35.42 | 13.05 | 22.37 | 0.63 |
| 500 | 27.87 | 12.97 | 14.90 | 0.53 |
从表3可以看出,和未充氢预处理的金属氧化物阳极相比,钛基体充氢后的氧化物阳极的各类伏安电量均明显增大,表明充氢预处理增大了氧化物阳极活性位点的数量。当充氢电流密度≤ 250 mA/cm2时,随着充氢电流密度的增加,无论是q
表4 钛基体未充氢和经不同电流密度充氢后制备的氧化物阳极的双电层电容和形貌因子
Table 4
| I / mA·cm-2 | Cdl / μF·cm-2 | φ |
|---|---|---|
| Uncharged | 5.31 | 0.133 |
| 50 | 5.75 | 0.144 |
| 100 | 7.97 | 0.199 |
| 250 | 8.27 | 0.207 |
| 500 | 8.05 | 0.201 |
由表4可以看出,钛基体充氢处理后,氧化物阳极的形貌因子和双电层电容均增大,表明钛基体充氢处理增大了氧化物阳极的表面粗糙度和电化学活性表面积。当充氢电流密度≤ 250 mA/cm2时,Cdl和φ会随着充氢电流密度的增加而升高,表明随着充氢电流密度的增加,阳极具有更大的电化学活性表面积,因而具有更高的电催化活性。但是当充氢电流密度达到500 mA/cm2时,Cdl和φ会有所减小。上述结果表明,循环伏安测试获得的变化规律与EIS测试结果相一致。
开展了金属氧化物阳极在3.5%NaCl溶液中的动电位极化测试,阳极极化曲线如图9所示。从图9可以看出,所有的金属氧化物阳极均呈现类似的阳极极化行为,随着极化电位的升高,电流密度先是快速增大,然后缓慢增加。这是因为当阳极电位达到析氯电位以上后,氧化物阳极表面发生析氯反应,导致电流密度随电位升高快速增大。随着电位的进一步升高,除了发生析氯反应外,还会发生析氧副反应,同时由于强烈的气体析出,使得阳极表面被气泡所覆盖,表面电化学反应受到一定的抑制,从而显著增大了阳极的极化率,导致电流密度增加缓慢[4, 7]。和钛基体未充氢的氧化物阳极相比,充氢预处理的氧化物阳极的极化曲线均明显右移,意味着钛基体充氢处理增大了金属氧化物阳极的电催化活性。在同样析氯电位下,随着充氢电流密度的增加,阳极的析氯活性逐渐增大。但充氢电流密度达到500 mA/cm2时,阳极析氯活性会有所降低但依然大于未充氢的阳极。不同氧化物阳极电催化活性的变化规律和阳极的电化学活性表面积的变化一致,表明增大氧化物阳极的电化学活性表面积可增强阳极的析氯电催化性能。
图9

图9
钛基体未充氢和经不同电流密度充氢后制备的氧化物阳极在3.5%NaCl溶液中的动电位极化曲线
Fig.9
Potentiodynamic polarization curves measured in 3.5%NaCl solution for the oxide anodes on titanium substrates uncharged and charged with hydrogen at different current densities
2.2.3 氧化物阳极的电化学稳定性
图10
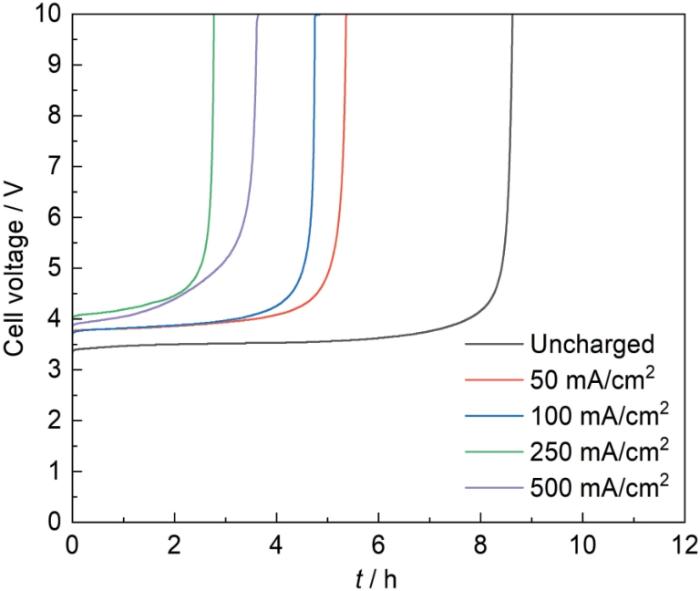
图10
钛基体未充氢和经不同电流密度充氢后制备的氧化物阳极在1 mol/L硫酸溶液中强化电解加速寿命测试(电流密度为1 A/cm2)时的槽压随时间的变化
Fig.10
Cell voltage vs. time curves recorded during accelerated life tests in 1 mol/L sulfuric acid solution at 1 A/cm2 for the oxide anodes prepared on titanium substrates uncharged and charged with hydrogen at different current densities
钛基体充氢处理对氧化物阳极寿命的影响与氧化物涂层的微观结构密切相关。随着充氢电流密度的增加,涂层表面出现更多大而深的裂纹,涂层的孔隙率增大(见表3),对基体的保护作用降低,使得电解质溶液更容易到达氧化物涂层与钛基体的界面,从而促进了钛基体表面的钝化,从而加速了氧化物阳极的失效。另外,随着充氢电流密度增大,钛基体表面所形成的氢化物增多,并且产生破碎和开裂,电化学测试也表明充氢处理后的钛基体耐蚀性明显降低,因此该氢化物层以及在热烧结过程中分解形成的产物层不能够对钛基体提供良好保护,结果导致氧化物阳极的寿命缩短。当充氢电流密度增加到500 mA/cm2时,阳极表面裂纹相较于充氢电流密度250 mA/cm2时有所减少,涂层的孔隙率有所降低,对钛基体保护作用有所提高,从而减缓了钛基体表面的钝化,使得阳极寿命有所增加。
3 结论
(1) 钛基体阴极充氢后表面形成了由TiH1.5和TiH2构成的氢化物层,随着充氢电流密度增大,表面氢化物增多并出现开裂。电化学测试表明,充氢后钛基体表面的膜电阻Rf和电荷转移电阻Rct减小,表面钝化膜的稳定性变差,钛基体的耐蚀性随着充氢电流密度的增大而降低。
(2) 钛基体充氢处理影响RuO2-IrO2-TiO2阳极表面微裂纹形貌,随着充氢电流密度的增加,涂层表面的粗裂纹逐渐增多,孔隙率增大,但当充氢电流密度增加到500 mA/cm2时,阳极表面裂纹和孔隙率又有所减少。XRD分析表明,氧化物涂层由金红石相的固溶体组成,但没有检测到氢化物的衍射峰,这可能是因为热烧结过程中钛基体表面的氢化物发生了分解转变。
(3) 钛基体充氢处理对RuO2-IrO2-TiO2阳极的电化学性能有明显影响,随钛基体充氢电流密度的升高,氧化物阳极的电化学活性表面积增大,析氯电催化活性增强,涂层内部的活性位点增多,但阳极的电化学稳定性降低。当充氢电流密度继续增加到500 mA/cm2时,则发生相反的变化,阳极的电催化活性有所降低,而强化电解寿命值较充氢电流密度为250 mA/cm2时增大。
参考文献
Development and application of electrolyzing seawater antifouling technique
[J].
电解海水防污技术的发展及应用
[J].
Electrocatalysis: Understanding the success of DSA®
[J].
Electrochemical behavior of the oxide-coated metal anodes
[J].
Performance study of RGO-CNTs hybrid material modified RuO2-IrO2-SnO2/Ti anode
[J].
RGO-CNTs杂化材料改性RuO2-IrO2-SnO2/Ti阳极的性能研究
[J].
Characterization and electrochemical behavior of Ti/TiO2-RuO2-IrO2-SnO2 anodes prepared by sol-gel process
[J]. J.
Effect of IrO2 loading on RuO2-IrO2-TiO2 anodes: A study of microstructure and working life for the chlorine evolution reaction
[J].
A comparative investigation of the corrosion behavior of RuO2-IrO2-TiO2 coated titanium anodes in chloride solutions
[J].
Surface characterization of RuO2‐IrO2‐TiO2 coated titanium electrodes
[J].
Structural analyses of RuO2-TiO2/Ti and IrO2-RuO2-TiO2/Ti anodes used in industrial chlor-alkali membrane processes
[J].
On the deactivation mechanism of RuO2-TiO2/Ti anodes prepared by the sol-gel procedure
[J].
Effect of oxalic acid etching on morphology and electrocatalytic activity of oxide anodes
[J].
IrO2--Ta2O5 oxide anode was prepared by coatingoxide on titanium substrate which hasbeen pretreated by sandblast and oxalic acid etching. ESEM,polarization curve and EIS indicated that the corrosion rate of thesubstrate and the loading of the oxide coating (i.e. Ir content)increase with etching time at the initial stage and thendecrease. The oxide coatingon the substrate pretreated by suitable etching pretreatment is of auniform and compact surface which has large electrochemical activesurface area, fine electrocatalyticactivity for oxygen evolution and high stability.
草酸浸蚀对氧化物阳极形貌及电催化性能的影响
[J].钛基体涂覆铱钽氧化物阳极制备过程中对基体喷砂并进行不同时间的草酸浸蚀。环境扫描电镜、析氧极化曲线、电化学阻抗谱测试表明, 草酸对钛基体的腐蚀速度及涂层载量(Ir含量)随腐蚀时间的延长先增大后减小。经过适当时间酸蚀处理后的基体上制备的氧化物阳极均匀致密, 其电化学活性表面积大, 析氧电催化活性提高, 并具有较长的使用寿命。
Origin of ohmic losses at Co3O4/Ti electrodes
[J].
A highly stable Ti/TiH x /Sb-SnO2 anode: preparation, characterization and application
[J].
Mn3O4@C micro-flakes modified Ti/TiH2/β-PbO2 anode for accelerating oxygen evolution reaction in zinc electrowinning
[J].
Formation mechanism of hydride precipitation in commercially pure titanium
[J].Since titanium has high affinity for hydrogen and reacts reversibly with hydrogen, the precipitation of titanium hydrides in titanium and its alloys cannot be ignored. Two most common hydride precipitates in α-Ti matrix are γ-hydride and δ-hydride, however their mechanisms for precipitation are still unclear. In the present study, we find that both γ-hydride and δ-hydride phases with different specific orientations were randomly precipitated in the as-received hot forged commercially pure Ti. In addition, a large amount of the titanium hydrides can be introduced into Ti matrix with selective precipitation by using electrochemical treatment. Cs-corrected scanning transmission electron microscopy is used to study the precipitation mechanisms of the two hydrides. It is revealed that the γ-hydride and δ-hydride precipitations are both formed through slip + shuffle mechanisms involving a unit of two layers of titanium atoms, but the difference is that the γ-hydride is formed by prismatic slip corresponding to hydrogen occupying the octahedral sites of α-Ti, while the δ-hydride is formed by basal slip corresponding to hydrogen occupying the tetrahedral sites of α-Ti.
Effect of electrochemical hydrogen charging on defect structure in titanium
[J].
Mechanical properties and hydrogen diffusion analysis of titanium alloy microstructure
[J].
钛合金微结构力学性能和氢扩散分析
[J].
Electrochemical and semiconducting properties of thin passive film formed on titanium in chloride medium at various pH conditions
[J].
Surface characterization of the commercially pure titanium after hydrogen charging and its electrochemical characteristics in artificial seawater
[J].
Effect of hydrogen precharging on mechanical and electrochemical properties of pure titanium
[J].
Effect of flowing seawater on corrosion characteristics of passivation film on TA2 pure-Ti pipes
[J].
流动海水冲刷下TA2纯钛管路钝化膜腐蚀特性研究
[J].TA2纯钛是新一代船舶海水管路材料,在海水冲刷下管路钝化膜失效会导致管路耐久性下降。研究海水冲刷对不同表面处理TA2纯钛腐蚀行为的影响对于评估TA2纯钛腐蚀和防护具有重要意义。采用动电位极化、电化学阻抗和Mott-Schottky分析等电化学测试方法研究了两种表面处理TA2纯钛在流动海水中的耐冲刷腐蚀性能,并对腐蚀后的试样进行腐蚀形貌观察和腐蚀产物分析。结果表明,5 m/s以内,海水流速的变化对TA2纯钛表面钝化膜的耐蚀性影响较小。电位升高时,在流动海水中钛合金表面钝化膜出现短暂的溶解现象,但很快就会进行再钝化修复,对材料的耐蚀性并未产生明显影响。相比于表面钝化处理试样,抛光状态试样在流动海水冲刷下阴极极化存在极限扩散特征,这主要是由于钝化状态试样表面已经形成了钝化膜,其对氧的消耗少于尚未形成钝化膜的表面抛光试样。抛光试样由氧传质速度控制的去极化发展慢,出现不随电位变化的极限电流密度。两种表面处理后的TA2纯钛钝化膜在海水中均只呈现n型半导体特征,且极化测试后材料表面平整,未出现明显局部腐蚀。
Preparation of ultrafine RuO2-IrO2-TiO2 oxide particles by a sol-gel process
[J].
Interfacial properties of oxides with technological impact in electrochemistry
[J].
“Inner” and “outer” active surface of RuO2 electrodes
[J].
Electrochemical cell design for the impedance studies of chlorine evolution at DSA® anodes
[J].
Novel porous molybdenum tungsten phosphide hybrid nanosheets on carbon cloth for efficient hydrogen evolution
[J].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}