



多数金属 (合金) 在自然环境中的腐蚀是电化学过程。因此,金属腐蚀的检测与研究大多是基于电化学方法[1]。传统电化学测试技术,包括稳态测试技术、暂态测试技术及电化学阻抗测试技术等,因操作简单、灵敏度高以及可实时获得电极反应过程动力学信息等优点,成为金属腐蚀研究中十分重要的工具[2],但也存在明显的局限性。传统电化学测试技术是以整个金属电极为测试对象、以电信号为刺激和/或检测手段,只能获得整个金属电极的电极电位、电流等电学信息。其中,电流密度是电极反应速率的直接量度,常用于确定整个电极反应的动力学参数。然而,金属的电化学腐蚀过程不仅涵盖了金属电极与腐蚀介质之间的电子转移,还涉及离子等物质的化学反应和传输历程。对于多组元材料,传统电化学测试技术测得的电流是各元素得失电子的总和,不仅包括目标元素反应,还包括其它平行反应,即便是纯金属的电化学反应,测得的电流也是金属阳极反应和H2、O2等阴极反应耦合后的结果。因此,传统电化学测试技术很难直接提供单一元素、单个阳极或阴极电化学反应过程的动力学信息,而这些信息对于深入剖析金属电极的微观腐蚀机制尤为重要。
为此,科技工作者往往借助X射线光电子能谱 (XPS)、Mossbauer谱、椭圆偏振以及二次离子质谱 (SIMS)、Fouier红外光谱 (FTIR) 等表面分析技术,移位获得金属电极表面元素成分和结构信息,以弥补电化学测试技术的缺陷。与此同时,电化学测试技术的上述缺陷也催生了各种电化学测试技术与表面分析技术联用的原位分析技术的研制和发展,如电化学测试技术与激光拉曼光谱联用技术使原位捕获电化学腐蚀过程中电极与腐蚀介质界面处不同组元的成分、结构变化成为可能,电化学测试技术与扫描探针测量技术联用使原位捕获电化学腐蚀过程中电极与腐蚀介质界面处的形貌变化成为可能,实现了腐蚀金属电极过程在分子级水平的微观原位研究,大大提高了金属腐蚀研究的深度和广度[3~14]。
尽管如此,上述技术仍无法满足电化学腐蚀理论研究的需要。金属的电化学腐蚀过程不仅涵盖了金属电极与腐蚀介质之间的电子转移,还涉及离子等物质的化学反应和传输历程。无论是何种形式的电化学腐蚀,腐蚀过程中必会有一部分金属以离子的形式进入到腐蚀介质中,而表面分析技术仅能追踪到电极表面不同组元的成分、结构变化,无法直接提供腐蚀介质本体的成分变化信息。因此,准确测定腐蚀介质本体的成分变化对深入开展电化学腐蚀理论研究至关重要。若能将电化学测试技术与元素成分分析手段相结合,该技术将对电化学腐蚀理论研究做出与上述联用技术同等重要的贡献。在众多元素成分分析手段中[15~17],电感耦合等离子体原子发射光谱 (ICP-OES) 是最适于腐蚀介质中腐蚀产物元素成分分析的技术之一,这是因为ICP-OES具有如下性能优势[18]:
(1) ICP-OES采用液体进样,可以直接对腐蚀介质中的腐蚀产物元素成分进行定性和定量分析;
(2) ICP-OES分析速度快,可在1 min内同时完成几十个元素的测定,所以可快速获得电化学控制下腐蚀介质中元素成分变化的实时数据;
(3) ICP-OES不仅能够测定元素周期表中除放射性元素、气体元素以及卤族元素外的绝大多数元素 (73种),而且还具有较高的灵敏度和较低的检出限,对于大多数元素而言,只要其质量浓度高于1~10 μg·L-1就可以有效检出;
(4) 仪器的动态线性范围宽,其工作曲线的直线范围可达4~5个数量级,可对主、次、痕量元素成分进行同时测定;
(5) ICP-OES耐盐度高,可分析固体含盐量高达30%的盐溶液,受腐蚀介质成分限制小;
(6) ICP-OES受基体干扰小,这得益于等离子体在高温条件下能够破坏所有的分子键并将其转化为原子的特点。
因此,若能将电化学测试技术与ICP-OES联用,实现在金属电极在发生电化学反应过程中对腐蚀介质中金属电极各合金元素含量变化的实时监测,进而计算出各种合金元素的实时溶解速率,再结合腐蚀介质中各种合金元素的变化规律以及金属电极的电化学参数的变化规律,可以更深入地剖析金属电极的微观腐蚀机制。
1 电化学-电感耦合等离子体原子发射光谱联用技术简介
1.1 电感耦合等离子体原子发射光谱
原子发射光谱,顾名思义是根据样品中受激发的原子跃迁回基态或更低能级所发射的电磁辐射 (光子) 来进行定量和定性分析的,其中元素的类型与发射谱线的波长有关,而元素的质量浓度与其特征谱线的强度有关[19]。处于基态的原子跃迁到激发态需要有激发光源,用于原子发射光谱的激发光源主要有:直流电弧、低压交流电弧、高压火花源、以及电感耦合等离子体 (ICP) [20]。ICP是一种利用电磁感应产生的无极放电等离子体,与其它激发光源相比,ICP的主要优势在于其工作时所产生的高温 (7000-8000 K) 能够使绝大多数样品中的绝大多数元素 (包括难熔元素) 高效的蒸发、原子化和激发,同时具有很好的重现性,现已成为原子发射光谱 (OES) 的主要激发源[18]。
以ICP为激发光源的OES被称为ICP-OES。ICP-OES出现于上世纪60年代,并于70年代迅速发展,商品化的ICP-OES仪主要由ICP发生器和OES光谱仪两大部分组成,ICP发生器包括:进样系统、等离子体炬管以及RF高频发生器;OES光谱仪包括分光器、检测器以及相关的电子数据系统[20]。ICP-OES进行测定时,液态样品可直接注入ICP中,而固态样品则需要用酸溶解转变成溶液后才可注入。
1.2 电化学-ICP-OES联用装置
图1

目前,已报道的用于电化学-ICP-OES的电化学流动池主要有两种。图2为Ogle和Weber设计的电化学流动池[21, 22]。该流动池为三电极两腔室结构,工作电极和辅助电极被渗析膜分隔在两个腔室,其作用是可避免两腔室溶液的大量混合。在工作电极室,暴露于电解液中的工作电极的表面面积由O形环的几何尺寸确定,其最终曝露面积为0.52 cm2,工作电极所在腔室的体积为0.27 cm3,电解液的流速的可调范围为2~12 mL·min-1,由位于上游的蠕动泵控制。工作时,电解液从池体底部进入,在流经金属电极表面时并与金属电极发生反应,随后携带所有来自金属电极的溶解产物从池体顶部流出,流入位于下游的ICP-OES仪中进行元素质量浓度检测。近年来,其它研究小组[15, 23]也提出了类似结构的电化学流动池。但值得指出的是,此类流动池有3个明显的局限性[24]:流动池的内腔体积一旦确定就无法改变;流动池的内腔结构导致溶液死体积较大;相对较大的内腔体积增加了工作电极表面流体的不均匀性。
图2

图3
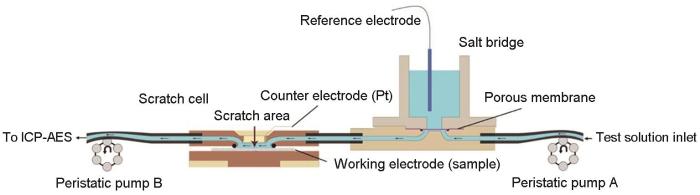
为了弥补上述电化学流动池的局限性,最近,中科院金属所腐蚀电化学研究团队[26]设计了一种新型电化学流动池 (图4),该池体主要由电解池模块、聚四氟乙烯垫片和工作电极模块三个部分组成。在电解池模块中,分别设有溶液进样通道和出样通道;3 M Ag/AgCl参比电极 (E=+0.208 V vs. NHE,25 ℃) 放置于工作电极的正对面;铂丝辅助电极 (0.1 mm,99.997%) 置于电解池的出样通道处,其目的是防止辅助电极上的反应产物对工作电极造成污染。聚四氟乙烯垫片 (厚度100 µm) 的中心为椭圆形孔洞,通过加紧固件夹于电解池模块和工作电极模块之间,构成小体积的储液槽。储液槽的体积可根据实际需要,增加或较少相应的垫片数量来调整。与Ogle和Weber设计的流动池[21]不同,该流动池将参比电极与工作电极置于同一腔室,有效降低了溶液电阻。
图4

1.3 电化学-ICP-OES的测试原理
利用电化学-ICP-OES进行测试时,蠕动泵首先以适当的速度将新鲜的电解质溶液不断地输送到电化学流动池中,建立相对稳定的电化学体系;而后利用电化学综合测试系统对电化学流动池中金属电极的电化学参数进行控制;与此同时,在蠕动泵的推动下,流动池中的电解质溶液携带腐蚀产物被推送至位于下游的ICP-OES进行目标元素的质量浓度检测[21]。若金属电极各合金元素在电化学流动池中主要发生如下的电化学反应:
假设某一瞬态t,流动池内金属电极组分M的溶解速率为vM (t),则金属电极组分M在流速为
式中,CM (t)为金属电极组分M在流动电解质中的质量浓度 (g·L-1),vM (t)为金属电极组分M的溶解速率 (g·s-1),f为电解质的流速 (L·s-1)。将
电解液流速f是电化学流动池的实验参数,是已知量。如不考虑流体在流动池和管线中停留而变宽等因素,则金属电极组分M在流动电解质中的质量浓度CM (t)可由位于电化学流动池下游的ICP-OES直接测得,因此通过
2 电化学-ICP-OES在金属腐蚀研究中的应用
电化学-ICP-OES同时测量金属电极反应过程中光谱信号和电信号的能力,不但可以提供金属电极表面各合金元素的瞬时溶解速率信息,当将这些数据与电化学数据 (比如外测电流) 相关联时,还可以精准的确定阴极半反应速率,金属表面膜的生成和溶解速率,以及反应的电子转移数等,为精准理解电极反应过程提供宝贵的定量数据[27, 28]。近年来电化学-ICP-OES在腐蚀领域的应用十分活跃,涵盖了多种材料,多种腐蚀介质,与多种电化学测试方法相结合,研究内容也涉及了很多方面。研究对象包括了纯金属 (纯铜[29]、纯铝[30]、纯镁[31~33]等)、合金 (不锈钢[21, 22]、铝合金[30, 34, 35]、铜合金[36]等)、金属中间相 (Al2Cu, Al7Cu2Fe, Al2CuMg)[37]、磷酸盐涂层[38]等。腐蚀介质涵盖了NaCl[23, 25, 31, 33]、Na2SO4[31]、NaOH[35]、HNO3[35]、H2SO4[37],Na2HPO4 /H2NaPO4[33]等。电化学测试方法用到了开路电位测试[25, 38]、恒电位/电流极化[23, 29, 36]、动电位扫描[36]、交流阻抗测试[34, 39]、恒电位阶跃[31~33]等。研究内容涉及了金属的阳极溶解[21, 31, 33]、金属的阴极溶解[30]、金属颗粒的脱落[35]、金属表面氧化膜的形成和溶解[35]、合金选择性溶解[22, 36, 37]等。
研究利用电化学-ICP-OES不仅可以获取金属电极的溶解速率、阴极的反应速率、有非可溶性产物的生成速率等详细的金属电极反应相关信息,还可验证元素的溶解价态。
2.1 元素溶解速率的测定
利用电化学-ICP-OES可以直接测量材料在开路状态下的溶解速率-时间/电位曲线。例如,如图5所示,Ogle研究组[38]利用电化学-ICP-OES实时测量了钢表面的磷酸盐转化涂层在0.1 mol/L NaOH中的溶解速率以及开路电位随时间的变化。t<0时,电解液绕过金属电极直接进入ICP-OES进行质量浓度检测,此时y轴所对应的为NaOH水溶液中原有元素的质量浓度,即空白值。t=0以及t>0时,NaOH溶液流过磷酸盐涂层表面与其发生反应。可以看出,在初始反应阶段,Zn和P的溶解速率迅速上升,P的溶解速率略高于Zn的溶解速率。随后,Zn和P的溶解速率迅速降低,但P比Zn的溶解速率的降低的更快。Mn的溶解速率也呈先升高后降低的趋势,但其最大速率比Zn和P的溶解速率低好几百倍。这个例子很好的证明了利用电化学-ICP-OES可以直观的看到金属电极中各元素的溶解速率随时间的变化,并且证明了该方法具有较低的检出限以及较宽的工作曲线线性范围,能够满足高、低含量元素的同时测定。
图5

2.2 阴极反应速率或不可溶腐蚀产物的测定
金属材料与腐蚀溶液的反应过程中可能会有非可溶性产物生成,例如阴极析氢、吸氧或滞留在金属电极表面的氧化物或氢氧化物。虽然电化学-ICP-OES仅能测得溶解到腐蚀介质中那部分腐蚀产物的质量浓度,但非可溶性产物的生成量可通过电荷平衡计算来间接量化。利用电化学-ICP-OES可同时测得金属电极在发生电化学反应过程中的电化学电流密度 (Ie) 以及溶解到腐蚀溶液中的各元素的溶解速率 (vMi )。根据vMi,可计算出总的阳极溶解电流 (IƩ):
当Ie=IƩ时,表明溶解反应的Faraday效率为100%,所有电化学电流全部用于生成可溶性产物;当Ie≠IƩ时,表明存在阴极反应或有不溶物生成,其电流密度 (IΔ) 可根据电荷/质量平衡公式间接量化:
式中,当IΔ>0且阴极电流密度可忽略不计时,IΔ为生成氧化物的电流密度 (Iox);当IΔ<0且非可溶性产物的生成可忽略时,IΔ为阴极反应电流密度 (Ic)。例如,图6为利用电化学-ICP-OES测得的AA6061中基体元素 (Al) 以微量元素 (Si 1.17%,Mg 0.68%) 在NaCl溶液中阴极极化下的溶解曲线,其目的是计算阴极电流 (OH-) 与Al溶解之间的化学计量比[30]。如图7所示,2000 s<t<3000 s对应的是样品表面的预活化步骤;t=3000 s时,对样品施加-1.5 V vs. SCE的电位进行阴极极化。在铝的阴极极化过程中,根据电荷平衡,阴极反应电流密度 (Ie) 可以用外测电流密度 (
图6
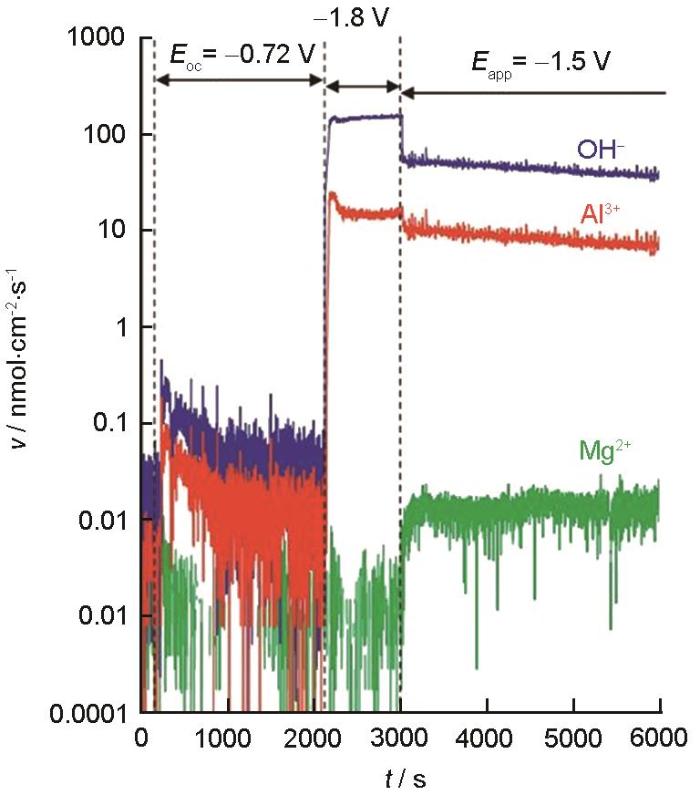
图7

2.3 元素的溶解化学计量数的确定
ICP的高温能够破坏样品的所有分子键并将其转化为原子,所有样品一旦进入等ICP中,其分子信息 (元素的氧化状态和分子形态) 就会丢失。因此,ICP-OES无法直接测量元素的溶解价态,但却可以对电化学-ICP-OES数据进行质量/电荷平衡运算间接求得。例如,当反应M→M n++ne-是唯一反应,且具有100%的Faraday效率时,ICP-OES测得的等效溶解电流密度 (IM (t)) 等于恒电位仪测得的Faraday电流密度Ie(t),即
式中,nM 为M在溶解过程中转移电子的化学计量数。如果采用n=nM 计算而得的IM (t)与Ie(t)相等,则表明M的溶解价数为nM。下面以纯镁为例[31],进一步解释这个计算。图7为纯镁在Na2SO4水溶液中一系列阳极恒电位脉冲下 (从左到右-1.4、-1.3、-1.2、-1.0、-0.8、-0.5和-0.3 V vs. Ag/AgCl) 的电化学-ICP-OES溶解曲线,黑色实线为假设n=2计算出的Mg的等效溶解电流密度 (
3 电化学-ICP-OES的关键技术
显然,将电化学测试技术与ICP-OES联用是研究金属电极腐蚀动力学过程的强有力手段。但若要将二者成功联用,还需解决如下几项关键技术问题:
3.1 电化学流动池的设计
在整个电化学-ICP-OES测试系统中,电化学流动池是决定测试结果可靠性的关键因素之一。不合理的流动池设计会导致流动池内部不同位置处溶质的扩散速度不同,从而形成浓度梯度,溶质不仅包括金属阳离子的浓度,还包括参与电化学反应的其他物质,比如Cl-、H+、OH-、O2,酸碱度等[24]。此外,流动池的内部结构、物理容积、电解液体积/工作电极面积比例均会对测试结果产生影响,是流动池设计不可忽略的关键要素。
3.2 工作参数的选择
作为电化学流动池和ICP-OES共同的工作参数,电解液流速是决定二者能否成功联用的另一重要因素。对于ICP-OES而言,增加流速能够增大待测元素在等离子体焰中的质量浓度,提高发射强度,同时对等离子体焰还有冷却作用,可降低发射背景,提高信噪比。但若流速过大,易形成大颗粒雾滴,使背景噪音增加,雾化器的雾化效率变差,还会使样品消耗量增加。对于电化学-ICP-OES联用系统而言,当金属电极的溶解速率一定时,电解液的流速越快,其对应的由ICP-OES测得的质量浓度就越小,由此可见联用系统的灵敏度与电解液流速成反比,增加流速会导致联用系统的灵敏度降低;此外,增加电解液流速还会增加流体在电极表面的不均匀性,产生成分梯度,最终影响测试结果的准确性[24]。除了对联用系统检测性能的影响之外,电解液流速也会在金属电极反应速率和反应机理的研究中起关键作用,特别是当某些电极反应受扩散控制的时候,电解液流速会直接影响金属电极的腐蚀行为[24]。因此电解液流速的合理选择,对提高联用系统的灵敏度,改善联用系统的时间分辨率、提高流体在电极表面的均匀性以及保证测试结果的可靠性非常重要。
3.3 光谱信号与电信号的同步性
4 结论与展望
新技术的进步和数据分析手段的完善,促进了元素分析测试技术在金属腐蚀研究领域研究中的广泛应用。电化学-ICP-OES结合了电化学检测技术的优势与ICP-OES的检测限低、动态线性范围宽及多元素同时测定的优点,能够在金属电极与腐蚀介质反应的过程中,实时原位监测从金属电极上溶解下来的绝大多数种类的物质,对材料和电解质溶液均无特殊要求,并且样品制备和定量过程简单易行,这将有助于金属材料的腐蚀行为和腐蚀机理研究的深入。迄今,电化学-ICP-OES在金属腐蚀研究方面已取得了长足的进步,各种新的分析方法已逐步建立并在实际样品的研究中得以应用,随着对金属腐蚀研究的重视度的提高以及分析技术和数据处理方法的不断完善,电化学-ICP-OES的应用前景将更加广阔。
参考文献
Research progress of application of electrochemical corrosion technology
[J].
腐蚀电化学技术应用研究进展
[J].
Influence of Ni, Mo, and Cr on pitting corrosionof steels studied by Raman spectroscopy
[J].
Thermally oxidized Inconel 600 and 690 nickel-based alloys characterizations by combination of global photoelectrochemistry and local near-field microscopy techniques (STM, STS, AFM, SKPFM)
[J].
Use of local electrochemical methods (SECM, EC-STM) and AFM to differentiate microstructural effects (EBSD) on very pure copper
[J].
In situ Raman spectroscopic identification of rust formation in Evans' droplet experiments
[J].
Progress in corrosion science at atomic and nanometric scales
[J].
Investigation of passive film properties and pitting resistance of AISI 316 in aqueous ethanoic acid containing chloride ions using electrochemical impedance spectroscopy(EIS)
[J].
Analysis of the composition of the passive film on iron under pitting conditions in 0.05 M NaOH/NaCl using Raman microscopy in situ with anodic polarisation and MCR-ALS
[J].
Local corrosion behavior of additive manufactured AlSiMg alloy assessed by SEM and SKPFM
[J].
In situ Raman spectroscopy and electrochemical techniques for studying corrosion and corrosion inhibition of iron in sodium chloride solutions
[J].
In situ and operando AFM and EIS studies of anodization of Al 6060: influence of intermetallic particles
[J].
Underpaint corrosion of zinc-coated steel sheet studied by in situ raman spectroscopy
[J].
SKPFM measured Volta potential correlated with strain localisation in microstructure to understand corrosion susceptibility of cold-rolled grade 2205 duplex stainless steel
[J].
In situ identification and quantification in a flow cell with AAS downstream analytics
[J].
Coupling of a high throughput microelectrochemical cell with online multielemental trace analysis by ICP-MS
[J].
Investigating the passivity and dissolution of a corrosion resistant Mg-33at.%Li alloy in aqueous chloride using online ICP-MS
[J].
Inductively Coupled Plasma/Optical Emission Spectrometry
[A].
Anodic dissolution of 304 stainless steel using atomic emission spectroelectrochemistry
[J].
Atomic emission spectroelectrochemistry applied to dealloying phenomena: I. The formation and dissolution of residual copper films on stainless steel
[J].
In-situ optical emission spectrometry during galvanostatic aluminum anodising
[J].
On the time resolution of the atomic emission spectroelectrochemistry method
[J].
Investigating ion release using inline ICP during in situ scratch testing of an Mg-Li(-Al-Y-Zr) alloy
[J].
New insight into the negative difference effect in aluminium corrosion using in-situ electrochemical ICP-OES
[J].
Atomic Emission Spectroelectrochemistry: a new look at the corrosion, dissolution & passivation of complex materials
[J].
Atomic emission spectroelectrochemistry: real-time rate measurements of dissolution, corrosion, and passivation
[J].
Atomic emission spectroelectrochemistry (AESEC) is a relatively novel technique that gives real-time elemental dissolution rates for a material/electrolyte combination, either reacting spontaneously or with electrochemical polarization. This methodology gives direct insight into questions such as how specific elements of an alloy interact with one another, or how specific additives in a surface treatment solution will affect different alloying elements or different phases. This paper discusses AESEC instrumentation and presents the basic quantitative relationships between the electrochemical and spectroscopic measurements. A wide range of applications are used to illustrate these relationships including the surface pretreatment of aluminum alloys (etching and deoxidation) and the passivation of Fe-Cr and Ni-Cr alloys. The focus is on the use of in-line inductively coupled plasma atomic emission spectroscopy (ICP-AES), although a brief discussion of similar techniques using in-line inductively coupled mass spectroscopy (ICP-MS) is included.
The anodic dissolution of copper alloys: pure copper in synthetic tap water
[J].
On the cathodic dissolution of Al and Al alloys
[J].
The anodic dissolution of Mg in NaCl and Na2SO4 electrolytes by atomic emission spectroelectrochemistry
[J].
The pH dependence of magnesium dissolution and hydrogen evolution during anodic polarization
[J].
Mg dissolution in phosphate and chloride electrolytes: insight into the mechanism of the negative difference effect
[J].
Refining anodic and cathodic dissolution mechanisms: combined AESEC-EIS applied to Al-Zn pure phase in alkaline solution
[J].In this work, the use of atomic emission spectroelectrochemistry (AESEC) coupled to electrochemical impedance spectroscopy (EIS) is presented as a method of revealing dissolution mechanisms. To illustrate the method, the dissolution kinetics of Al cations from an Al-Zn pure phase (Zn-68 wt.% Al) was investigated in an alkaline solution. In the cathodic potential domain, a nearly direct formation of dissolved Al3+ was observed, while in the anodic potential domain the Al dissolution occurred by migration across a ZnO/Zn(OH)2 film. It was demonstrated that this methodology can be applied to a nonstationary system during a potentiostatic experiment for a lower Al content phase (Zn-22 wt.% Al). The nature of the charge transfer mechanisms depended on the applied potential and could be identified by comparing the direct current and alternating current faradaic yield using AESEC-EIS.
In-situ monitoring of alloy dissolution and residual film formation during the pretreatment of Al-Alloy AA2024-T3
[J].
Interactions between elemental components during the dealloying of Cu-Zn alloys
[J].The elemental dissolution of Cu-Zn alloys was investigated as a function of Zn content ranging from 0 to 45 wt%. Atomic emission spectroelectrochemistry (AESEC) was utilized to directly monitor Cu2+ and Zn2+ release and oxide growth as function of time during potentiodynamic experiments. It was determined that Cu dissolution undergoes a simultaneous mechanism of Cu2O formation and Cu2+ release. The addition of Zn in Cu-Zn alloy does not measurably change the dissolution mechanism of Cu2+ and the rate of aqueous Cu2+ was only dependent on the potential. Zn dissolution was however blocked by the formation of a Cu(0) film which shifted the Zn dissolution in the anodic direction. (c) 2018 Elsevier Ltd.
Dealloying of Al2Cu, Al7Cu2Fe, and Al2CuMg intermetallic phases to form nanoparticulate copper films
[J].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}